The Medical Device Regulation (MDR), Regulation (EU) 2017/745, defines strict requirements for the conformity assessment of medical devices in the EU. Our experts guide you through the entire process and support your market access with structured assessment procedures – reliably, efficiently and in compliance with standards.
Why MDR certification?
market access
Access to the European market with over 500 million potential users for your medical devices.
Global recognition
MDR compliance is increasingly recognised as a quality standard outside the EU as well.
MDR conformity assessment with Berlin Cert
As a Notified Body, we offer comprehensive services as part of our conformity assessment in accordance with:
Assessment procedures:
Review of technical documentation for all risk classes
QM system audits in accordance with MDR requirements
Product testing and type testing in accordance with the specific annexes of the MDR
Review of clinical evaluations and clinical evidence
Leading expertise in the rapidly developing market of software as a medical device and AI applications
Accompanying support:
Regular review of your post-market surveillance measures
Evaluation of change notifications, e.g. in the case of product modifications
Support with post-market surveillance
Information on regulatory developments (without compromising our neutrality as a Notified Body)
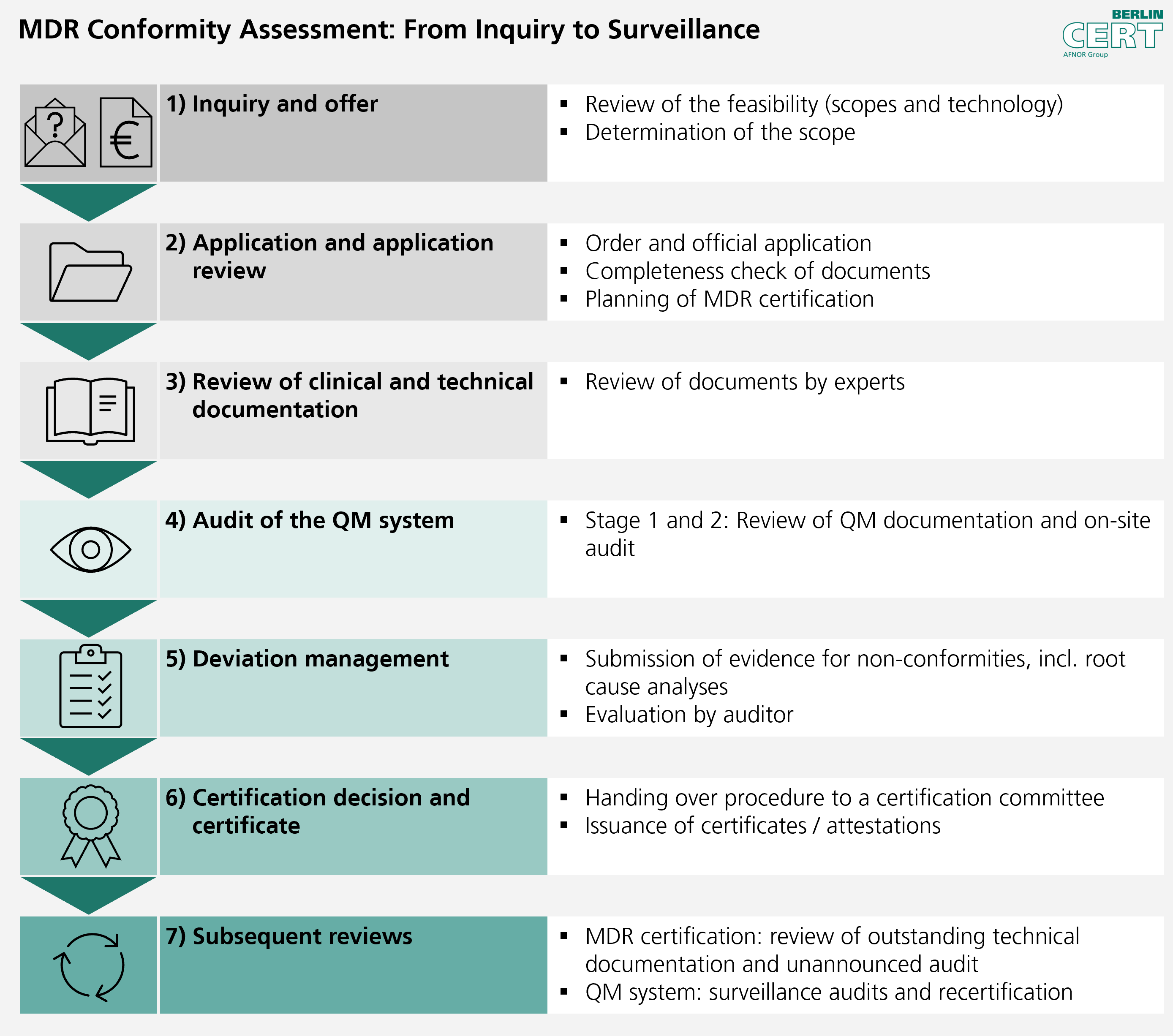
Based on the short questionnaire you have completed and the product attachments, we will check whether your request fits within our scope. If your request can be implemented by Berlin Cert, we will provide you with a transparent quote.
To prepare the quote, we need the completed short questionnaire and product attachments. We are happy to answer any questions you may have regarding the necessary information in online sessions.
2) Application and application review
The customer places an order and submits an official application. Once the application fee has been received, the completeness of the documents is checked within four weeks and MDR certification is scheduled.
Please ensure that all documents are complete at the time of application. Rejections or withdrawals of applications are communicated to EUDAMED and are visible to other Notified Bodies.
3) Review of technical documentation and clinical evaluations
Our technical experts evaluate your technical documentation for compliance with the fundamental safety and performance requirements of the MDR. Particular attention is paid to reviewing the clinical evaluation, risk management and biocompatibility.
4) Audit of the QM system
We conduct a comprehensive audit of your quality management system, which ideally is already certified according to ISO 13485. In doing so, we evaluate the implementation and effectiveness of all quality-related processes required for the manufacture of the medical device.
5) Deviation management
Any deviations or deficiencies identified are clearly documented and communicated. You are given the opportunity to implement corrective measures and provide evidence of this. We check and evaluate the effectiveness of these measures.
6) Certification decision and certificate
After successfully evaluating all aspects, the notified body makes the certification decision. If the result is positive, the EU certificate of conformity is issued, which allows you to affix the CE marking and distribute your product on the European market.
7) Subsequent monitoring
After initial certification, regular surveillance audits and inspections are carried out to ensure that compliance is maintained on an ongoing basis.
As a notified body, Berlin Cert certifies:
Efficiency:
Structured processes for rapid assessment procedures – also available as fast track
Technical expertise:
Our auditors have many years of experience in the medical device sector.
Transparency:
Clear communication throughout the entire process and transparent decisions
Neutrality:
Consistent compliance with the prohibition on consulting by notified bodies/certification bodies
Berlin Cert offers certifications in accordance with EU Regulation 2017/745 (MDR) for Annexes IX, X, XI(A) and XI(B) for medical devices in classes Ir, Im, IIa, IIb and III in the following scopes:
MDA 0202: Active non-implantable products for imaging procedures using non-ionising radiation
MDA 0203: Active non-implantable products for monitoring vital physiological parameters
MDA 0204: Other active non-implantable products for monitoring and/or diagnosis
MDA 0302: Active non-implantable devices using non-ionising radiation
MDA 0305: Active non-implantable devices for stimulation or inhibition
MDA 0306: Active non-implantable devices for extracorporeal circulation, for the administration or removal of substances and for haemopheresis (restriction: Excluding active devices intended to administer medicinal products, body fluids or other substances to the body and/or remove them from the body, where this procedure poses a potential risk, taking into account the nature of the substances concerned, the part of the body concerned and the type of application (Rule 12, Annex VIII, Regulation (EU) 2017/745)
MDA 0307: Active non-implantable respiratory devices
MDA 0309: Active non-implantable ophthalmic products
MDA 0310: Active non-implantable ear, nose and throat products
MDA 0311: Active non-implantable dental products
MDA 0312: Other active non-implantable products for surgery
MDA 0313: Active non-implantable prostheses, rehabilitation products and patient positioning and transport products
MDA 0315: Software
MDA 0316: Medical gas supply systems and their parts
MDA 0317: Active non-implantable products for cleaning, disinfection and sterilisation (restriction: cleaning and disinfection only)
MDA 0318: Other active non-implantable products
MDN 1205: Non-active non-implantable products for orthopaedics and rehabilitation
MDN 1214: General non-active non-implantable products used in healthcare and other non-active non-implantable products
MD 1301: Devices for monitoring non-vital physiological parameters
MD 1302: Devices for monitoring vital physiological parameters
Repair, maintenance and installation of medical devices (in the scopes as above)
Manufacturers of custom-made products in the fields of orthopaedics and orthopaedic shoe technology, rehabilitation technology and medical supply stores
Trade in medical devices
Transport of medical devices
Preparation of technical documentation for active medical devices, non-active orthopaedic products and non-active instruments (this includes the preparation of clinical evaluations)
The MDR divides medical devices into four risk classes:
Class I: Low risk (e.g. wheelchairs, bandages) Class IIa: Medium risk (e.g. dental fillings, hearing aids)
Class IIb: Increased risk (e.g. ventilators, X-ray equipment)
Class III: High risk (e.g. heart valves, implants)
The higher the risk class, the more intensive the continuous monitoring of products and organisation by the notified body and the authorities.