Die Medical Device Regulation (MDR), Verordnung (EU) 2017/745, definiert strenge Anforderungen an die Konformitätsbewertung von Medizinprodukten in der EU. Unsere Experten begleiten Sie durch den gesamten Prozess und unterstützen mit strukturierten Bewertungsverfahren Ihren Marktzugang – zuverlässig, effizient und normkonform.
Warum eine Zertifizierung nach MDR?
Marktzugang
Zugang zum europäischen Markt mit über 500 Millionen potenziellen Nutzern für Ihre Medizinprodukte.
Globale Anerkennung
Die MDR-Konformität wird zunehmend als Qualitätsstandard auch außerhalb der EU anerkannt.
MDR-Konformitätsbewertung mit Berlin Cert
Als Benannte Stelle bieten wir Ihnen im Rahmen unserer Konformitätsbewertung nach MDR umfassende Dienstleistungen:
Bewertungsverfahren:
Überprüfung technischer Dokumentationen für alle Risikoklassen
QM-System-Audits nach den Anforderungen der MDR
Produktprüfungen und Baumusterprüfungen gemäß den spezifischen Anhängen der MDR
Überprüfung von klinischen Bewertungen und klinischen Nachweisen
Führende Expertise auch im sich rasant entwickelden Markt von Software as a Medical Device und KI-Anwendungen
Begleitende Unterstützung:
Regelmäßige Sichtung Ihrer Maßnahmen zur Überwachung nach der Inverkehrbringung
Bewertung von Änderungsmitteilungen, z.B. bei Produktmodifikationen
Unterstützung bei der Nachmarktüberwachung
Informationen zu regulatorischen Entwicklungen (ohne Einschränkung unserer Neutralität als Benannte Stelle)
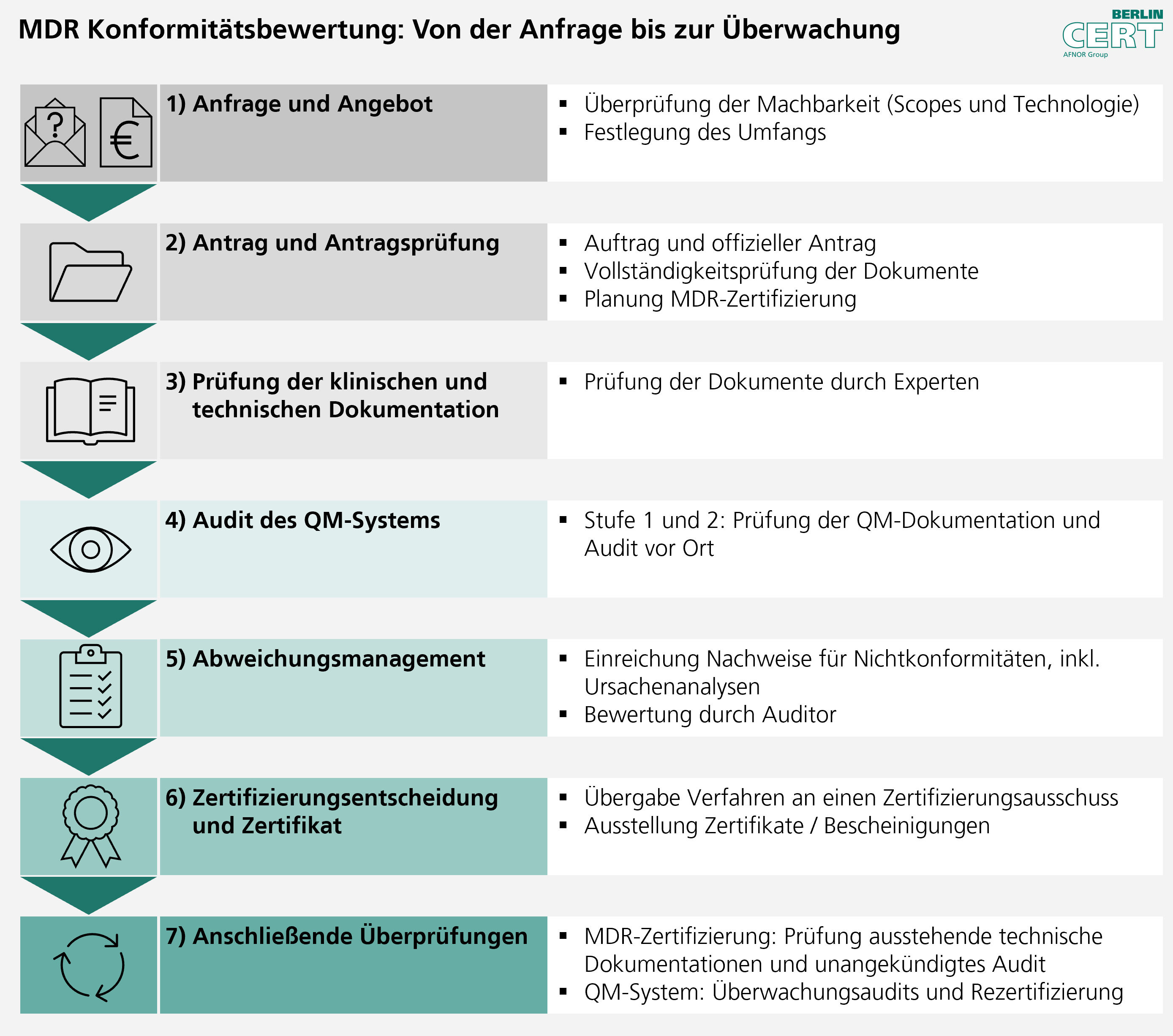
Auf Basis des durch Sie ausgefüllten Kurzfragebogens und der Anhänge Produkt überprüfen wir, ob Ihre Anfrage zu unseren Scopes passt. Ist Ihre Anfrage durch die Berlin Cert umsetzbar, erstellen wir Ihnen ein transparentes Angebot.
Zur Angebotserstellung benötigen wir den vollständig ausgefüllten Kurzfragebogen und Anhänge Produkt. Gerne klären wir Ihre Fragen bzgl. der notwendigen Informationen in Online-Sessions.
Dauer: typischerweise 1-2 Monate
2) Antrag und Antragsprüfung
Der Kunde erteilt einen Auftrag und stellt einen offiziellen Antrag. Nach Eingang der Antragsgebühr wird die Vollständigkeit der Dokumente innerhalb von vier Wochen überprüft und die MDR-Zertifizierung geplant.
Bitte stellen Sie sicher, dass zum Antragszeitpunkt alle Dokumente vollständig vorliegen. Ablehnungen oder Widerrufe von Anträgen werden an EUDAMED übermittelt und sind für andere Benannte Stellen einsehbar.
Dauer: typischerweise 1-1,5 Monate
3) Prüfung der technischen Dokumentation und klinischen Bewertungen
Unsere Fachexperten bewerten Ihre technische Dokumentation auf Übereinstimmung mit den grundlegenden Sicherheits- und Leistungsanforderungen der MDR. Besonderes Augenmerk liegt auf der Prüfung der klinischen Bewertung, dem Risikomanagement und der Biokompatibilität.
4) Audit des QM-Systems
Wir führen ein umfassendes Audit Ihres Qualitätsmanagementsystems durch, das idealerweise bereits nach ISO 13485 zertifiziert ist. Dabei bewerten wir die Implementierung und Wirksamkeit aller qualitätsrelevanten Prozesse, die für die Herstellung des Medizinprodukts erforderlich sind.
5) Abweichungsmanagement
Identifizierte Abweichungen oder Mängel werden klar dokumentiert und kommuniziert. Sie erhalten die Möglichkeit, Korrekturmaßnahmen zu implementieren und nachzuweisen. Wir prüfen und bewerten die Wirksamkeit dieser Maßnahmen.
6) Zertifizierungsentscheidung und Zertifikat
Nach erfolgreicher Bewertung aller Aspekte trifft die Benannte Stelle die Zertifizierungsentscheidung. Bei positivem Ergebnis wird das EU-Konformitätszertifikat ausgestellt, das Ihnen erlaubt, die CE-Kennzeichnung anzubringen und Ihr Produkt im europäischen Markt zu vertreiben.
7) Anschließende Überwachung
Nach der Erstzertifizierung folgen regelmäßige Überwachungsaudits und -prüfungen, um sicherzustellen, dass die Konformität kontinuierlich aufrechterhalten wird.
Als Benannte Stelle zeichnet die Berlin Cert aus:
Effizienz:
Strukturierte Prozesse für zügige Bewertungsverfahren - auch als Fast Track
Fachliche Expertise:
Unsere Auditor*innen verfügen über langjährige Erfahrung im Medizinprodukte-Sektor
Transparenz:
Klare Kommunikation über das gesamte Verfahren und nachvollziehbare Entscheidungen
Vertraulichkeit
Der Schutz vertraulicher Informationen steht für uns an oberster Stelle.
Neutralität:
Konsequente Einhaltung des Beratungsverbots von Benannten Stellen / Zertifizierungsstellen
Berlin Cert bietet Zertifizierungen nach der EU-Verordnung 2017/745 (MDR) jeweils für die Annexe IX, X, XI(A), XI(B) für Medizinprodukte der Klassen Ir, Im, IIa, IIb und III in folgenden Scopes an:
MDA 0202: Aktive nichtimplantierbare Produkte für bildgebende Verfahren mit nicht-ionisierenden Strahlen
MDA 0203: Aktive nicht implantierbare Produkte zur Überwachung von vitalen physiologischen Parametern
MDA 0204: Sonstige aktive nichtimplantierbare Produkte zur Überwachung und/oder Diagnose
MDA 0302: Aktive nichtimplantierbare Produkte mit nicht-ionisierenden Strahlen
MDA 0305: Aktive nichtimplantierbare Produkte zur Stimulation oder Hemmung
MDA 0306: Aktive nichtimplantierbare Produkte für extrakorporale Kreisläufe, zur Verabreichung oder Entfernung von Stoffen und zur Hämopherese (Einschränkung: Ohne aktive Produkte, die dazu bestimmt sind, Arzneimittel, Körperflüssigkeiten oder andere Stoffe an den Körper abzugeben und/oder aus dem Körper zu entfernen, wenn diese Vorgehensweise unter Berücksichtigung der Art der betreffenden Stoffe, des betreffenden Körperteils und der Art der Anwendung eine potenzielle Gefährdung darstellt (Regel 12 Anhang VIII Verordnung (EU) 2017/745))
MDN 1214: Im Gesundheitswesen verwendete allgemeine nichtaktive nichtimplantierbare Produkte und sonstige nichtaktive nichtimplantierbare Produkte
MD 1301: Geräte zur Überwachung von nicht vitalen physiologischen Parametern
MD 1302: Geräte zur Überwachung von vitalen physiologischen Parametern
Reparatur, Instandhaltung und Installation von Medizinprodukten (in den Scopes wie oben)
Hersteller von Sonderanfertigungen in den Bereichen Orthopädie- und Orthopädieschuhtechnik, Rehatechnik und Sanitätshäuser
Handel mit Medizinprodukten
Transport von Medizinprodukten
Erstellung technischer Dokumentationen für aktive Medizinprodukte, nicht-aktive orthopädische Produkte und nicht-aktive Instrumente (dies schließt die Erstellung klinischer Bewertungen mit ein)
Die MDR teilt Medizinprodukte in vier Risikoklassen ein:
Je höher die Risikoklasse, desto intensiver erfolgt die kontinuierliche Überwachung von Produkten und Organisation durch die Benannte Stelle und die Behörden.