FAQ's zur MDR Zertifizierung
MDR Konformitätsbewertung
Die Medical Device Regulation (MDR, Verordnung (EU) 2017/745) ist seit Mai 2021 vollständig in Kraft und ersetzt die bisherige Medizinprodukterichtlinie 93/42/EWG (MDD). Als EU-Verordnung gilt sie unmittelbar in allen Mitgliedstaaten und stellt erhöhte Anforderungen an die Sicherheit, Leistung und Rückverfolgbarkeit von Medizinprodukten. Die MDR definiert umfassende Regelungen für die Herstellung, den Vertrieb und die Überwachung von Medizinprodukten mit dem Ziel, den Patientenschutz zu verbessern.
[14.07.2025]
Die Konformitätsbewertung ist ein strukturiertes Verfahren, bei dem geprüft wird, ob ein Medizinprodukt die grundlegenden Sicherheits- und Leistungsanforderungen der MDR erfüllt. Je nach Risikoklasse des Produkts sind unterschiedliche Verfahren vorgeschrieben, die von der Selbstzertifizierung (nur bei Klasse I) bis hin zu umfassenden Überprüfungen durch Benannte Stellen reichen. Das Ergebnis einer erfolgreichen Konformitätsbewertung ist die CE-Kennzeichnung, die den Marktzugang im Europäischen Wirtschaftsraum ermöglicht.
[14.07.2025]
Als Benannte Stelle führen wir den Konformitätsbewertungsprozess nach einem klar strukturierten Verfahren durch:
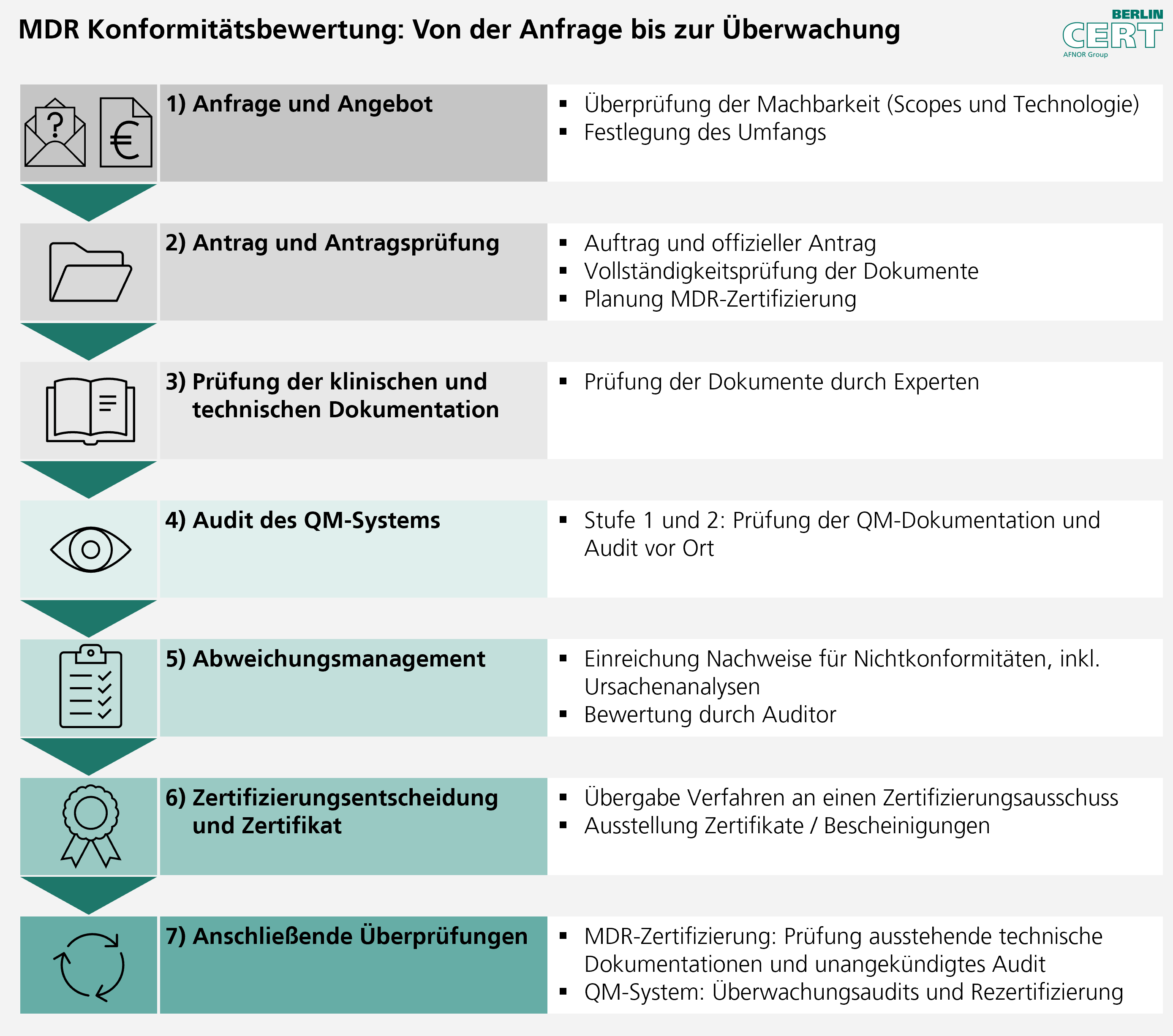
[14.07.2025]
Die EN ISO 13485 und die MDR stehen in einem engen Zusammenhang, ergänzen sich jedoch in ihren Anforderungen und Zielsetzungen.
Komplementäre Anforderungen
- EN ISO 13485: Definiert die Anforderungen an das Qualitätsmanagementsystem für Medizinprodukte
- MDR: Legt gesetzliche Anforderungen für den EU-Marktzugang fest
Die EN ISO 13485-Zertifizierung ist keine rechtliche Voraussetzung für die MDR-Konformität, bildet jedoch den anerkannten Stand der Technik für die Erfüllung der QM-Anforderungen der MDR. Ein nach EN ISO 13485 zertifiziertes Qualitätsmanagementsystem erfüllt bereits viele der in der MDR geforderten organisatorischen und prozessualen Elemente
Die MDR liefert die spezifische Ausgestaltung der in der EN ISO 13485 offen gehaltenen regulatorischen Anforderungen für den europäischen Wirtschaftsraum.
Unterschiede und Ergänzungen
Die MDR geht in einigen Bereichen über die Anforderungen der ISO 13485 hinaus:
- Spezifischere Anforderungen an die klinische Bewertung
- Detailliertere Regelungen zur Überwachung nach der Inverkehrbringung
- Umfassendere Transparenzanforderungen (EUDAMED)
- Strengere Regelungen für wirtschaftliche Akteure
[14.07.2025]
- Marktzugang: Legaler Zugang zum europäischen Markt mit über 500 Millionen potenziellen Nutzern für Ihre Medizinprodukte.
- Rechtssicherheit: Nachweis der Erfüllung aller gesetzlichen Anforderungen und damit Minimierung von Haftungsrisiken.
- Wettbewerbsvorteil: Differenzierung durch nachgewiesene Sicherheit und Leistungsfähigkeit in einem stark regulierten Markt.
- Vertrauen: Stärkung des Vertrauens von Patienten, Anwendern und Gesundheitseinrichtungen in Ihre Produkte.
- Globale Anerkennung: Die MDR-Konformität wird zunehmend als Qualitätsstandard auch außerhalb der EU anerkannt.
[14.07.2025]
Die Umstellung auf die MDR stellt viele Hersteller vor erhebliche Herausforderungen:
Dokumentation und Nachweise:
- Umfangreichere technische Dokumentation
- Höhere Anforderungen an klinische Daten
- Strengere Nachweise zur Biokompatibilität und chemischen Charakterisierung
Prozessanpassungen:
- Implementierung erweiterter Vigilanz- und Nachmarktsysteme
- Anpassung der Lieferkettenkontrolle
- Umsetzung der UDI-Anforderungen
Ressourcen:
- Höherer Zeit- und Kostenaufwand für die Konformitätsbewertung
- Bedarf an spezialisierten Fachkräften
- Längere Vorlaufzeiten für die Markteinführung
[14.07.2025]
